
What is Pelizaeus-Merzbacher Disease (PMD)?
The History of PMD

PMD Foundation board member, Carlos Labrada with his wife, Yezabel and their son Leo, who was diagnosed with PMD at 6 months of age.
In 1885, a German physician named Pelizaeus described five boys in a single family with involuntary oscillatory eye movement, spasticity in the limbs, very limited head and trunk control, and delay in cognitive development.
In 1910, another German physician named Merzbacher reexamined this family, which then included 14 affected individuals including two girls, and found that all affected family members shared a common female ancestor.
In addition, he noted that the disease was passed exclusively through the female line without father-to-son transmission. Pathological analysis of brain tissue from one affected individual showed that most of the central white matter generally lacked histochemical staining for myelin, the fat and protein based substance that acts as an insulator for nerve conductors in the Central Nervous System (CNS). The description of this family provides the clinical, genetic and pathological basis for Pelizaeus-Merzbacher Disease (PMD): an X-linked disorder of myelin classically characterized by nystagmus, spastic quadriparesis, ataxia and cognitive delay in early childhood.
“Individuals with PMD have little to no myelin - the insulating sheath around many nerve fibers.”
What is PMD?

Figure 1. Myelin is produced by cells in the brain called oligodendrocytes that myelinate the axons, which are projections of nerve cells. The cell membrane on the outside of an oligodendrocyte wraps around the axons of several nerve cells. Cell membranes of other oligodendrocytes also wrap the axon until there are sheaths of myelin covering the axons of each nerve cell. The myelin sheaths provides insulation for axons so that nerve impulses can be transmitted rapidly from neuron to neuron without leaking out. This is similar to how we use insulation on electric cords.
Pelizaeus-Merzbacher disease (PMD) and spastic paraplegia 2 (SPG2) are genetic diseases of the central nervous system (brain and spinal cord) caused by mutations of a gene called the proteolipid protein 1 gene (PLP1). They belong to a group of diseases of the white matter called leukodystrophies. The white matter, which is in the central part of the brain, is mostly made of a fatty substance called myelin. Myelin is produced by cells in the brain called oligodendrocytes (see Figure 1). The cell membrane on the outside of the oligodendrocyte wraps around axons forming a myelin sheath surrounding fibers that project from a different cell type, the neuron (nerve cell). The myelin sheath provides insulation for axons so that nerve impulses can be transmitted rapidly from neuron to neuron without leaking out. This is similar to how we use insulation on electric cords. In PMD, the myelin doesn’t form properly and some nerve impulses are slowed so they reach their target more slowly than normal, while some leak out and do not reach their target at all.
It is estimated that PMD affects 1 in 200,000 to 1 in 500,000 live births.
Diagnosis of PMD/SPG2 is usually made by a neurologist based on family history (when available) and clinical examination with the aid of magnetic resonance imaging (MRI). Molecular confirmation can be obtained by genetic testing for DNA variants (mutations) of the proteolipid protein 1 gene (PLP1) that include both DNA sequence variants and copy number changes (See Genetics of PMD section).
The clinical signs of PMD include:
Nystagmus (involuntary eye movements)
Hypotonia (decreased muscle tone)
Spasticity (stiffness or increased muscle tone; hypotonia progresses to spasticity)
Titubations (head-bobbing)
Stridor (high-pitched wheezing sound)
Sometimes vocal paralysis in severe cases
Feeding difficulties
Delayed developmental milestones
Ataxia of the limbs
Cognitive impairment
Sometimes seizures
Sometimes dystonic posturing (abnormal fixed posture)
Sometimes athetotic movements (involuntary writhing)
On magnetic resonance imaging (MRI), a diffuse pattern of hypomyelination (low amount of myelin) is seen in the white matter regions of the brain. When children with PMD are under the age of 1 or 2, they may not have a definitively abnormal MRI. This is because the brain is just becoming myelinated during this time. However, there are some changes in myelination seen on a MRI during this time that can suggest PMD or other leukodystrophy.

Figure 2. There is a wide spectrum of disease severities in PMD/SPG2. Disease severity ranges from the most severe connatal (or congenital) form that is present at birth in which patients never walk or talk to the least severe or spastic paraplegia 2 (SPG2) form in which patients develop the ability to walk and talk, but they decline usually beginning in the teen years.
There is a wide spectrum of disease severities in PMD/SPG2 (see Figure 2). Patients within a family are usually similarly affected, but there can be some variability. The types of PMD from most severe to least are:
Connatal PMD
Connatal means congenital or at birth, so the connatal type of PMD is observed very early in life. Patients never develop the ability to walk, use their upper extremities, or talk, but they may comprehend what is said. Although it can be found in the literature that connatal patients die in their first decade, care has improved and they often live longer, but lifespan is shortened.Classic PMD
This is the type of PMD that was originally described by Drs. Pelizaeus and Merzbacher (see History of PMD), and it is the most common type. Some patients develop the ability to walk with an assistive device (like a walker), and lose the ability as disease progresses. Often they can talk, but speech is dysarthric (slow and difficult to understand). Lifespan may be shortened, but patients with classic PMD have lived to their sixth or seventh decade.Complicated spastic paraplegia 2 (SPG2)
This is a milder form of PMD called by a different clinical name, SPG2, in which patients are able to walk with a spastic gait because of muscle tightness, but they lose that ability and use a walker and then a wheelchair as disease progresses. It is called “complicated” because these patients also have dysfunction of their autonomic nervous system (the part of the nervous system that supplies the internal body parts), like issues with their urinary bladder. These patients can talk, but speech may be slow. Lifespan may be normal.Pure spastic paraplegia 2 (SPG2)
This form is the same as complicated SPG2 but without the autonomic nervous system issues.
There is no cure for PMD.
Treatments involve management of symptoms and supportive therapies, including:
Medications for seizures and spasticity
Physical therapy and exercise
Occupational therapy
Speech therapy
Orthotics for spasticity and scoliosis management
Surgery for contractures and scoliosis
Gastrostomy (feeding tube placement) for severe dysphagia (swallowing difficulty)
Proper wheelchair seating
Special education
Assistive communication devices
Genetics Background Information
The cell is a basic unit that is able to perform the functions of life (see Figure 1). Cells have, among other parts, a nucleus where the chromosomes reside. The chromosomes encode information that tell the cell what to do and when to do it. Chromosomes are made of deoxyribonucleic acid (DNA) and the encoded information is in the DNA. The double helix of DNA spends most of its time spread out in the nucleus of cells, but when cells get ready to divide, the chromosomes condense, and we can see in the microscope that humans have 23 pairs of chromosomes (See Figure 2). The pairs are numbered by size from the largest, 1, to smallest, 22. In addition, there is a pair of sex chromosomes (XY in males and XX in females).
The main code in the DNA is the one for making proteins.
There are many types of proteins. Some are structural building blocks. Others are enzymes that allow chemical reactions to take place. Proteins must be made and transported to where they belong by the cell.
A protein is made of a string of amino acids (see Figure 3). There are 20 types of amino acids (for example, phenylalanine, leucine, serine, and cysteine are shown in Figure 3). Each protein has to have the right amino acids in the right places in the string for the protein to fold up properly and do what it is supposed to do in the cell.
How the genetic code works is shown in Figure 4. Generally speaking, the region of DNA along a chromosome that specifies one protein is called a gene. The code in the DNA specifies the linear order in which amino acids get added to make a specific protein.
There are four structural units or bases that make up the DNA code. The bases are called adenine, thymine, cytosine, and guanine, which we shorten to the letters A, T, C, and G. The DNA code for an amino acid is a 3-letter word (called a codon).
For example, the codon TTT in DNA tells the protein-making machinery of a cell to add the amino acid called phenylalanine to a growing protein. At the end of the string of codons along the DNA that specify a protein is a 3-letter word or codon that tells the protein-making machinery to stop adding amino acids to that protein, called a stop codon.
To understand one of the types of variants (mutations) that people with PMD can have, that is, splicing variants, you need to know about two intermediate steps between the DNA and protein.
In one intermediate step before the protein can be made, the DNA code has to be transcribed to another chemical called ribonucleic acid (RNA), which is similar to DNA but is single-stranded. Figure 4 shows the RNA that gets made in the step between the DNA code and the protein.
RNA is similar to DNA in that it is linear and has bases along its length. Three of the bases are the same as those in DNA (adenine, cytosine, and guanine) and the fourth, uracil, is similar to thymine. The RNA-making machinery of the cell reads a codon of DNA and adds the codon to the growing RNA, substituting U for T in the process called transcription.
In the second intermediate step before the protein can be made, another process called splicing occurs, in which portions of the RNA are cut out of the RNA and only the code in the remaining processed RNA is translated into protein. The parts of the RNA that get spliced out are called introns. The parts that remain in the RNA and code for the protein are called exons because we say that they are expressed by the cell. The process of splicing is shown in Figure 5. It is the code in the spliced RNA (called messenger RNA or mRNA) that actually gets “read” by the protein-making machinery in cells.
While the intermediate steps involving RNA between the DNA and the protein make it more complex and difficult to understand how the genetic code works and certainly made it much more challenging for scientists to discover the processes in the first place, they allow much more flexibility for the cell. For example, the RNA produced from one gene can be spliced in several different ways, so actually instead of one gene making one protein, it can make several related proteins with somewhat different functions because of alternative splicing.

Figure 1. A cell showing nucleus with chromosomes. The chromosomes are made of the DNA double helix all wound up and held together by proteins (shown in yellow). Between the two strands of the double helix are the bases (or letters A, T, G, and C) that are responsible for the genetic code. The bases on one side of the helix are bonded to the complementary bases on the other side of the helix. A always pairs with T, and G always pairs with C. A gene is a stretch of DNA that codes for a protein.

Figure 2. The karyotype of a normal person. The karyotype with chromosomes grouped by size shows that humans normally have 23 pairs of chromosomes. One chromosome of each pair came from the person’s mother and the other from his/her father. This is the karyotype of a male as there is an X and a Y.
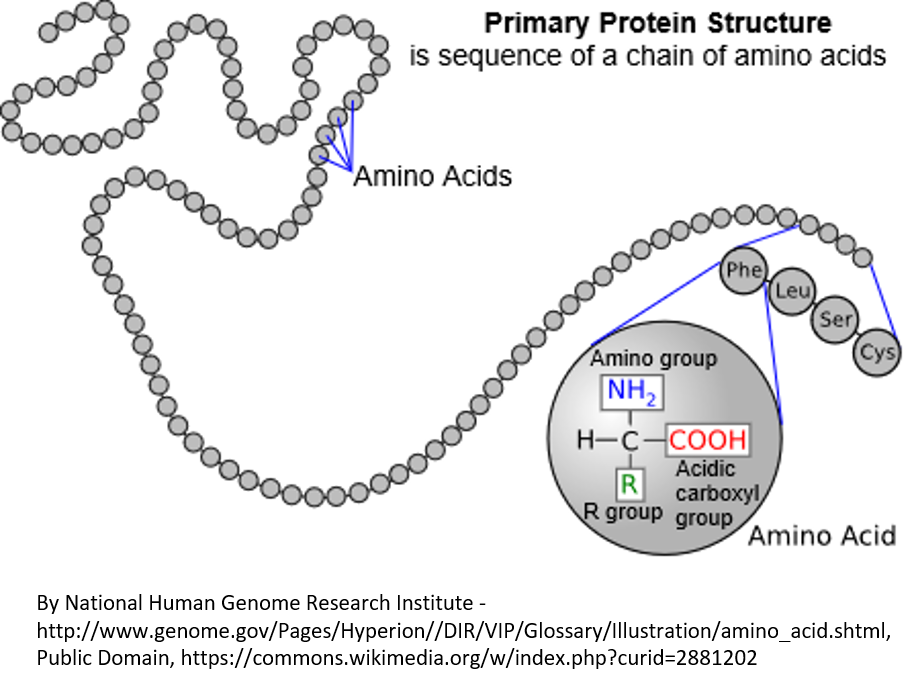
Figure 3. Proteins, which are coded by DNA, are made of a linear string of amino acids. In this diagram, the amino acids are represented by circles. There are 20 amino acids that can be used in various combinations to make proteins. In an expanded region of the protein, the amino acids phenylalanine, leucine, serine, and cysteine are shown. The three-letter standard abbreviations for these amino acids is shown in circles. There are also a one-letter standard abbreviations that are sometimes used. In the large circle, you can see the chemical elements that are the same for every amino acid, plus the R group that differs for each of the 20 amino acids and gives them their different properties.
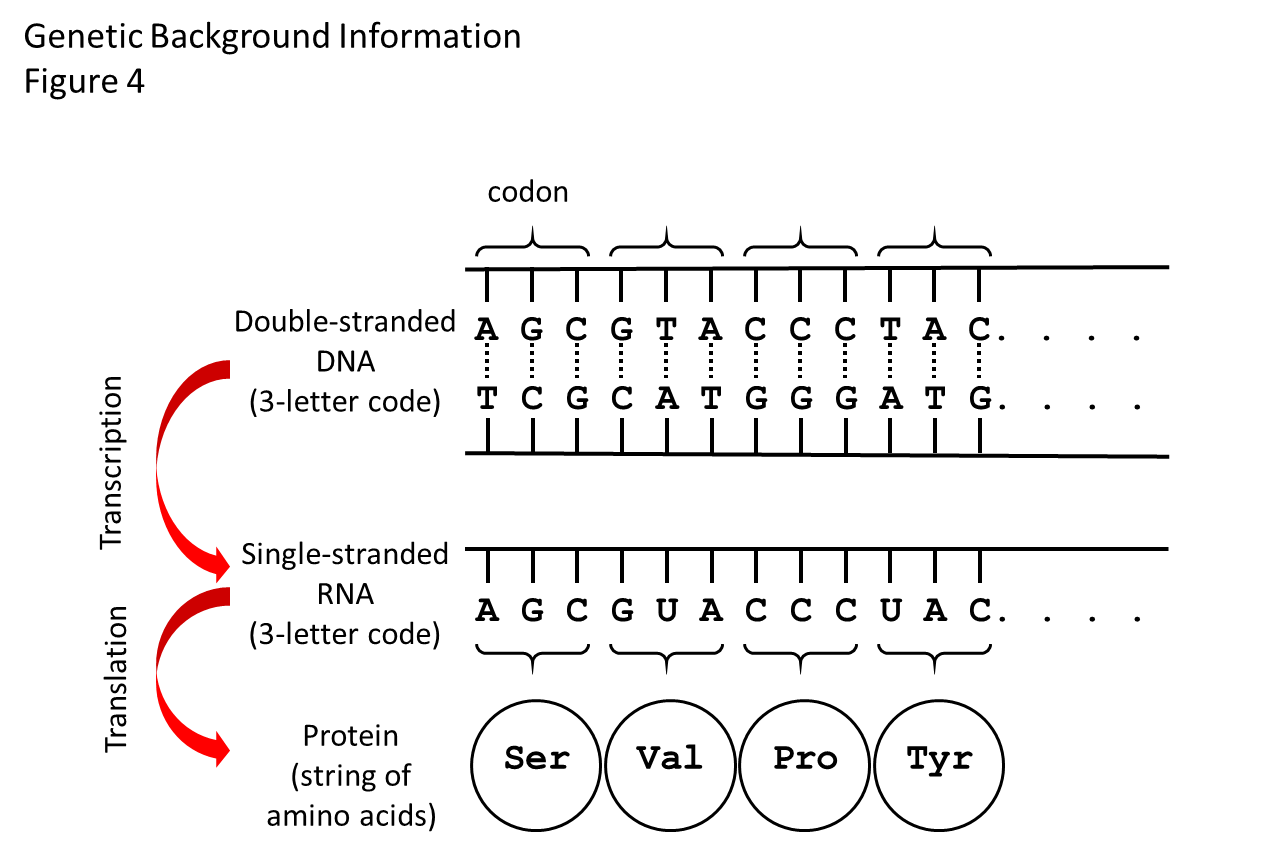
Figure 4. How the genetic code works. There is a flow of information from DNA to RNA to protein. The double helix of the DNA shown in this diagram is stretched out flat. Between the two strands of the double helix are the bases that make the 3-letter words (codons) of the genetic code. There are four bases: adenine (A), thymidine (T), cytosine (C), and guanine (G). The bases on the top strand are complementary to the bases on the bottom strand. A on one strand always pairs with T on the other, and C always pairs with G. The bases on one strand are weakly bonded to the bases on the other strand (shown by dashed lines), so that the strands of DNA can be zipped apart and the cellular machine that “reads” the DNA code and makes the RNA strand can get to the strand that gets copied. The RNA strand will have the same code as the DNA, except that it uses the base called uracil (U) instead of T. Another cellular machine then “reads” the RNA three-letter words one codon at the time and adds the specified amino acids one at the time to the growing protein. The dots mean “and so on,” as the code goes on to make a protein with many amino acids, while only 4 are shown here.
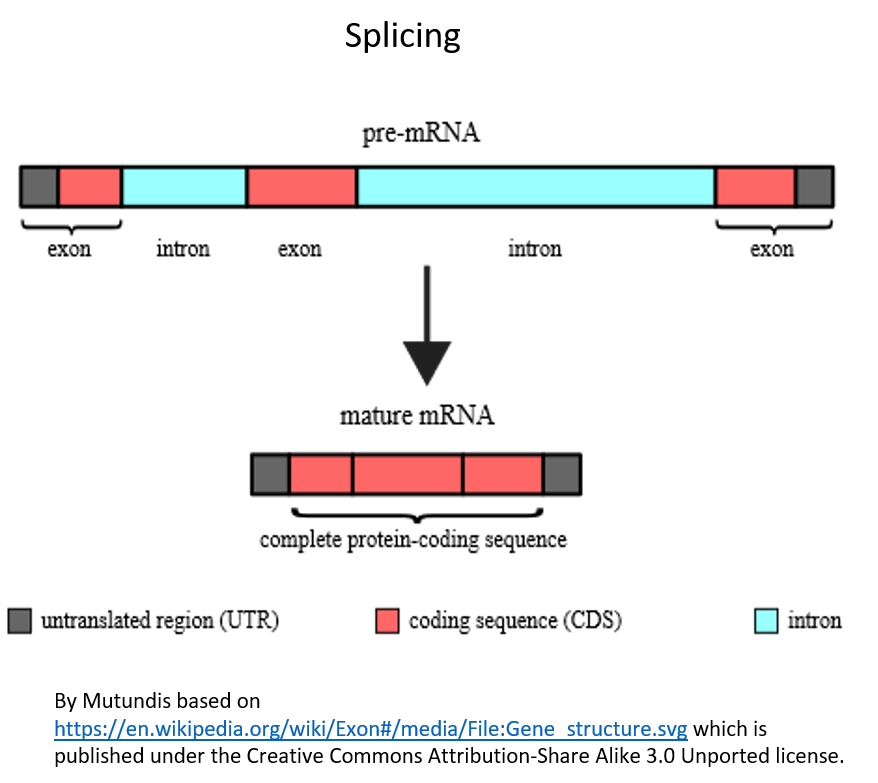
Figure 5. Splicing of the RNA. Splicing must occur so that only the expressed parts of the RNA (those that code for the protein) remain in the RNA. The introns (shown in blue) are spliced out and the exons (the parts that codes for the protein, shown in red) will remain.
Genetics of PMD/SPG2: X-chromosome inheritance
Variants (mutations) of the proteolipid protein 1 gene (PLP1) cause Pelizaeus-Merzbacher disease (PMD) and spastic paraplegia 2 (SPG2). Figure 1 shows the location of the PLP1 gene on the X chromosome.
The PLP1 gene provides the genetic code for a protein called proteolipid protein (PLP) to be made in cells of the central nervous system (brain and spinal cord) called oligodendrocytes. Oligodendrocytes are the cell type that make myelin, and PLP protein, which sits in the cell membrane of oligodendrocytes, is the major protein in myelin.
Figure 2 shows the amino acid structure of the PLP protein and how it spans the cell membrane four times.
Since the PLP1 gene is on the X-chromosome, PMD is an X-linked disease. This affects the inheritance pattern of PMD (see Figure 3).
The X and Y chromosomes determine sex. Males have one X and one Y, and females have two Xs. If a male has a mutated PLP1 gene on his X chromosome, he will have PMD. If a female has a mutated PLP1 gene on one of her X chromosomes, the normal PLP1 gene on her other X chromosome will compensate for it and she will usually not be affected. We call her a carrier because she carries the mutated PLP1 gene and can pass it to her offspring, but she is not affected.
Female carriers who mate with a normal male have a 25% chance of having an unaffected son, 25% chance of having an unaffected daughter, 25% chance of having a daughter who is a carrier, and 25% chance of having a son who has PMD.
Just like with rolling the dice, these chances are the same with every child the mother has, so if she already has one son who has PMD, her chances of having a son with PMD in the next pregnancy are still the same as they were for the first pregnancy, 25%. Males who have PMD do not usually have children, but some of the ones with the least severe form of PMD do. When a male with PMD mates with a normal female, there is a 50% chance of having a normal son, 25% chance of having a normal daughter, and 25% chance of having a daughter who is a carrier.
In some families, female carriers of PLP1 gene variants are affected with mild features of the condition, such as mild spasticity (muscle stiffness). These are usually the families in which variants in the males are not very severe, which seems counterintuitive.
A process called X-inactivation has been proposed as an explanation. In all cells in females, one of the X chromosomes is inactivated, meaning that the genes on this chromosome are silenced (they do not make protein).
Usually, X-inactivation is random from cell to cell, so half the time the X with a PLP1 variant is active and half the time it is not. In females where the variant causes severe disease in males, if the normal X chromosome is inactive and the X chromosome with the mutated PLP1 is active, the cell is sick and dies. That allows the cells with the normal active X to populate the brain. But in females where the variant causes milder disease in males, the cells with the active mutated PLP1 are sick but they don’t die, and there are enough “sick” cells to cause some symptoms.

Figure 1. The PLP1 gene is on the long arm (the p-arm) of the X chromosome. Giemsa banding (or G-banding) is a technique used to make a characteristic banding pattern appear on each of the chromosomes. The PLP1 gene is located in a band on the X chromosome called Xq22.3.

Figure 2. PLP protein sits in the cell membrane of oligodendrocytes. Like all other proteins, PLP is made of a string of amino acids. Each circle in the diagram with a letter inside represents an amino acid in the string that makes up the PLP protein. The one-letter abbreviations for amino acids are used here. M is for the first amino acid methionine; G is for glycine; L is for leucine; and another L for the next amino acid which is another leucine, etc. There is also a 3-letter abbreviation for each amino acid. There are 20 different amino acids. The dark circles with zig-zag lines represent the lipid bilayer that makes up the cell membrane. The amino acids that run through the lipid bilayer are in the oligodendrocyte cell membrane. Those that are above it are on the outside of the cell membrane, and those that are below it are on the inside of the cell in the cytoplasm, the substance that fills cells.

Figure 3. X-linked recessive inheritance. PMD has an X-linked recessive inheritance pattern. It is X-linked because the PLP1 gene is on the X chromosome. It is called recessive because if there is an affected copy of the gene on one chromosome and an unaffected copy on the X chromosome that is paired with it in a female, the unaffected copy is able to compensate for the affected copy and the female does not have PMD. This diagram of X-linked recessive inheritance shows that an unaffected father and a mother who is a carrier (has an unaffected copy of the PLP1 gene on one of her X chromosomes and an affected copy on the other) will have (shown left to right) the following probabilities for their offspring: ¼ (25%) unaffected son, ¼ (25%) unaffected daughter (not a carrier), ¼ (25%) carrier daughter, and ¼ (25%) affected son.
Genetics of PMD/SPG2: Types of variants in PMD patients
Genetics professionals now use the term “variant” instead of mutation for the changes that occur in the bases of DNA. The reason for this is that the word mutation had taken on the connotation of being pathogenic (detrimental; disease causing). However, some DNA changes are benign (not pathogenic), so another word was chosen for base changes that occur in DNA.
When a variant is discovered in a molecular testing lab, genetics professionals today apply guidelines produced by the American College of Medical Genetics to interpret the variant as benign, likely benign, likely pathogenic, pathogenic, or uncertain significance.
The variants that cause PMD/SPG2 can be:
Copy number variants
Duplication is the most common type of variant that causes PMD (See Figure 1). About 70% of PMD patients have a duplication. In duplications, the whole PLP1 gene plus other nearby genes on the X chromosome become duplicated, so there are two copies of the whole PLP1 gene and other genes instead of one. Presumably, too much PLP protein gets made in the brains and spinal cords of these patients.
Triplication (3 copies) and higher copy number changes are known to occur, but they are rare.
Complete and partial deletions of the PLP1 gene cause a small fraction of PMD cases (See Figure 2).Missense variants (see Figure 3A)
Missense variants occur when one base (letter) in one codon (3-letter word) of the genetic code is changed and the variant causes an incorrect amino acid to be added to the protein. (See Figure 3)Nonsense variants (Figure 3B)
Nonsense variants occur when a codon (3-letter word) is changed to a stop codon so that the growing chain of amino acids is prematurely terminated.Deletion or insertion of one or a few bases (Figure 3C)
When one or a few bases (letters) are deleted or inserted and the remaining bases are still read 3 letters at a time, there is a frameshift and the new sequence of bases codes for the wrong amino acids to be added until eventually a stop codon is reached and the growing chain of amino acids is terminated. When bases are inserted or deleted, the variant is sometimes called an indel (for insertion or deletion).Splicing variants (See Figure 4)
Splicing variants occur when there is a base change in a different code in the DNA from the one that specifies which amino acid is added in a protein. It is a change in the code that tells the splicing machinery of the cell where to splice the RNA, so the RNA is misspliced (see Figure 4).
Sometimes a part of the RNA that is supposed to be spliced out (the intron) does not get spliced out. Sometimes a part of the RNA that is not supposed to be spliced out (the exon) does get spliced out. In either of these cases, the amino acid chain in the PLP protein that gets made is not correct. In the PLP1 gene, there is an alternative splice that occurs normally leading to two related proteins being made from the gene, and some variants make this splice occur improperly so that the amounts of the two proteins that are made is incorrect.
The wide clinical spectrum of disease severity (see “What is PMD?” section) is reflected in the variety of types of variants that cause PMD/SPG2 (See Figure 5). Missense variants are usually misfolded and become stuck in a part of the cell where they are made (the endoplasmic reticulum) rather than being transported to the cell surface where they can become a part of the myelin. The more accumulation of PLP protein there is, the worse the myelin is and the more severe the disease is. The most severe variants, the connatal ones, are usually missense variants, but missense variants can cause symptoms across the severity spectrum, depending on where they occur in the gene. Those that occur in a part of the gene that is spliced out in one of the normal alternative proteins of the PLP1 gene and not the other can cause the mildest form, pure SPG2. Duplications usually result in classic PMD in the middle of the severity spectrum, but some patients with more severe and some with less severe disease have been reported. Triplications and higher copy number variants are on the connatal end of the spectrum. Complete deletion causes the milder complicated SPG2. It has been referred to as the null phenotype (null because there is no PLP protein; phenotype is another word for the clinical symptoms). Nonsense variants and splicing variants that occur as the first few amino acids are being added to the chain can also cause the null phenotype. Other nonsense variants can cause more severe symptoms if misfolded protein gets made.

Figure 1. The most common variant causing PMD is duplication. A duplication occurs when a part of the chromosome is doubled. Duplications can affect many genes in addition to PLP1.
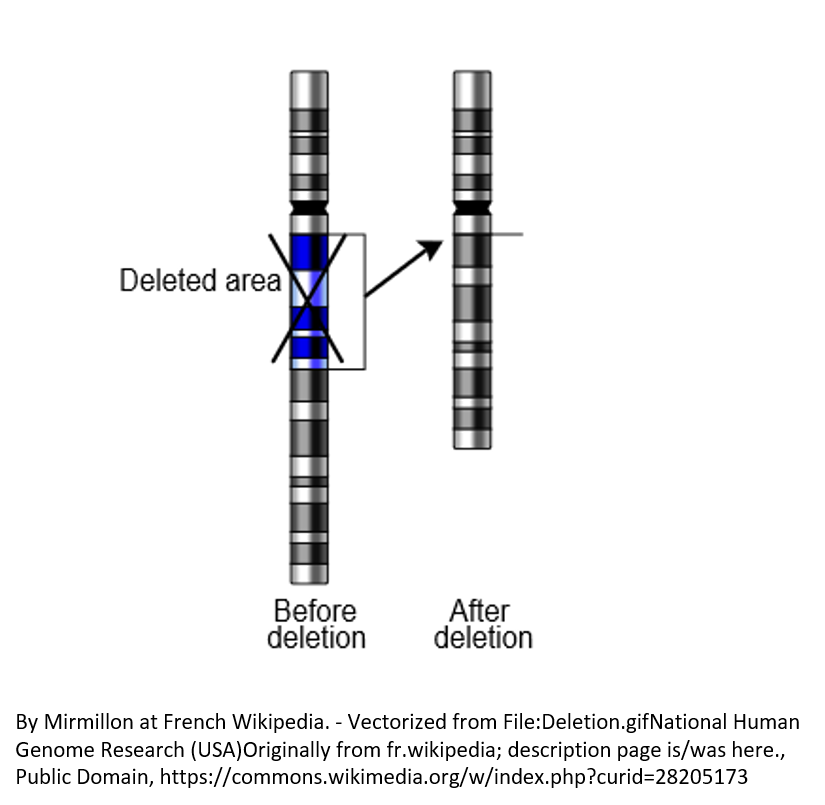
Figure 2. Rarely, complete deletion of a chromosomal region causes PMD.

Figure 3. Types of variants. A. A missense variant results in an amino acid change. B. A nonsense variant results in a stop codon and termination of the growing chain of amino acids. C. A deletion or duplication variant of one or several bases (in this case, a deletion of two bases) shifts the reading frame, so the wrong amino acids get added to the protein. What was originally the word GTA coding for Val is shifted to the word GCC, which also codes for Val, but then the next word is CTA coding for Leu rather than CCC coding for Pro. The remaining codons are frame shifted until a stop codon is reached by chance and then the growth of the amino acid chain is terminated. Addition of one or a few bases can have the same result as deletion.

Figure 4. Splicing variants. Splicing variants can cause exons to be skipped or introns to be retained in the RNA that gets translated to protein. It can also cause only parts of the protein to be made.

Figure 5. The wide clinical spectrum of disease severity (see “What is PMD?” section) is reflected in the variety of types of variants that cause PMD/SPG2.
Molecular testing for PMD/SPG2
Molecular testing is generally the next step after a patient is clinically diagnosed with a leukodystrophy that is suspected to be PMD or SPG2. Today, there are many labs and a variety of types of tests from which to choose. The Geneticist (MD) and Genetics Counselor are best professionals for determining what type of testing should be ordered. They are also the best for identifying an appropriate testing lab, and for explaining to patients and the family members what the test results mean and what their options are.
There are several types of tests available in clinical testing labs for identifying leukodystrophy variants in many genes using one test. These are useful when a doctor determines that a patient has one of the leukodystrophies, but does not know for sure which one.
Copy number variants (duplications and higher copy number; deletions) can be identified with a test called array comparative genomic hybridization (aCGH). In this test, the patient’s DNA is compared with normal DNA to find copy number variant regions across the genes in the genome. This test will detect PLP1 copy number variants in addition to copy number variants of other genes.
Variants of the DNA code (like missense, nonsense, insertion and deletion of one or a few bases, and splicing variants) can be detected by*:
exome sequencing in which the expressed parts of genes (those that code for the amino acids in the protein) are sequenced (sequencing means the order of bases or letters of the DNA code is determined)
genome sequencing in which most of the DNA is sequenced. This includes regulatory regions and parts that will get spliced out from the RNA
leukodystrophy panels in which the panel is designed to detect variants in the expressed regions of a selection of genes that are known or suspected to cause leukodystrophy
These tests will detect most sequence variants in PLP1. They also detect variants in other genes, so if the patient has a leukodystrophy other than PMD/SPG2, it could be detected with these methods.
When these global sequencing methods are used, trio testing will frequently be done. This means the patient, mother, and father will be tested at the same time. Having the parents’ results can help with the interpretation of variants found in the patient for pathogenicity. When a PLP1 variant is detected in the patient, the mother’s carrier status will be known if trio testing was done.
Targeted sequencing of only the PLP1 gene and methods that detect copy number changes only in PLP1 can be done when suspicion of PMD/SPG2 by clinical exam is great*. Targeted methods can also be used for testing of additional family members when a PLP1 variant is known in an affected family member.
Carrier detection (testing females to determine if they are carriers), prenatal testing (testing of a fetus before birth), and preimplantation genetic diagnosis (testing mother’s eggs prior to in vitro fertilization) require knowledge of the pathogenic variant in the family.
*Note: Pathogenic mutations are known to occur in intron 3 of the PLP1 gene, but not all of the tests used in molecular testing labs include the sequencing of intron 3. (See https://www.ncbi.nlm.nih.gov/pubmed/26125040)
Current Outlook for PMD
There is currently no cure for Pelizaeus-Merzbacher disease, nor is there a standard course of treatment. Treatment, which is symptomatic and supportive, may include medication for seizures and the stiffness or spasticity that most PMD patients have. Once a PLP gene mutation is identified in a family, it is possible to test family members for the mutation and to provide prenatal diagnosis for parents who have a risk of transmitting this disorder. The prognosis for those with Pelizaeus-Merzbacher Disease varies. Some mutations are more severe than others and may result in death during childhood, but some individuals can survive into their sixties. The course of the disorder is usually very slow, with some individuals reaching a plateau and remaining stable for years. However, some do worsen over time, for reasons to be investigated. A group of clinicians and researchers working on Pelizaeus-Merzbacher disease and PLP has recently been organized in the North America and in Europe to promote research to facilitate understanding of disease pathogenesis and development of specific treatments and, we hope, a cure.